![[實際案例分享] 以 HiFi 定序技術更精確地完成微菌叢分析與總體基因體定序 | PacBio](img/header.png)
|
⫹ 實際案例分享 ⫺ |
|
|
自 1670 年代微生物學之父 Antonie van Leeuwenhoek 以自製顯微鏡發現第一個微生物以來,時至今日我們已學習到唯有與無處不在的微生物建立良好平衡的共存關係,才能達到人類群體與整個地球生態環境共享健康 (One Health) 的理想狀態。近年來應運而生的「微生物體 (microbiome) 研究」即是為了釐清在特定場域中(如腸道、土壤)存在有哪些微生物,以及由這些微生物聚集組成的「微菌叢 (microbiota)」又將會如何與宿主和周遭生態環境產生互動及影響 [1]。
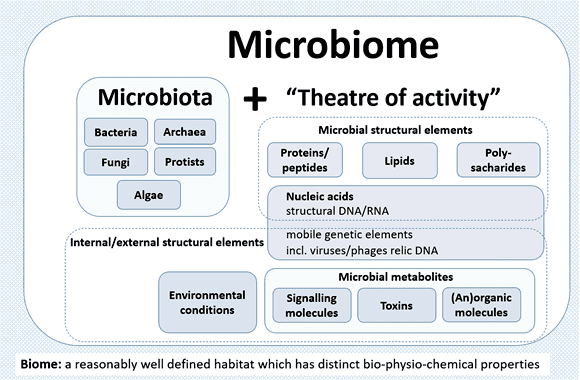
圖 1﹑由歐盟所贊助支持的 MicrobiomeSupport project 彙整了全球上百位微生物專家學者的意見,將「微菌叢 (microbiota)」與「微生物體 (microbiome)」這兩個名詞重新定義與詮釋,此共識的建立將有助於未來微生物體研究的標準化與成果整合。「微菌叢」意指特定環境下聚集存活的微生物,成員包含細菌、古菌 (archaea)、原生生物 (protist)、真菌 (fungi)、藻類 (algae)。而「微生物體」則是涵蓋微菌叢及其活動場景(包含微生物本身結構成分(例如遺傳物質、蛋白質、脂質、多醣體等)、外在環境條件、其他移動性遺傳元素(例如病毒、噬菌體、DNA 殘骸)、以及宿主或本身產生的代謝物等)。 IMAGE © Microbiome. 2020 Jun 30;8(1):103. Fig. 2 [1]. Pacific Biosciences 公司(簡稱 PacBio)所開發的 HiFi 定序技術, 在微生物體研究領域,能夠協助科研人員更精確地完成微菌叢分析和總體基因體定序與組裝 (metagenomic sequencing and metagenome assembly) 工作。以下就為您簡單地介紹幾個實際案例。
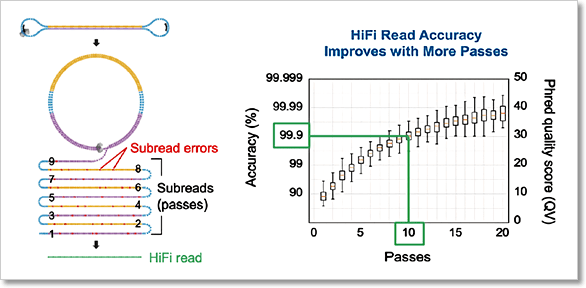
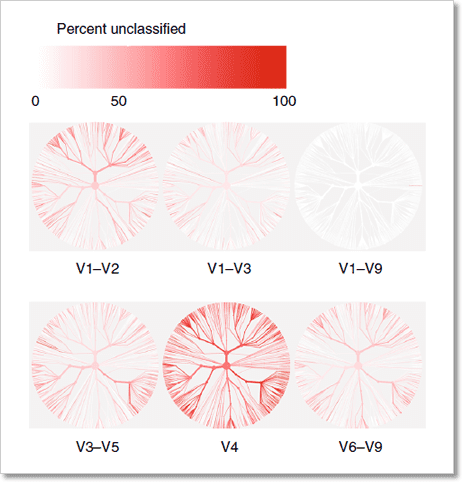
圖 2﹑高準確度的 HiFi 長讀取 (long-read) 定序技術。在進行 HiFi 定序時,待測 DNA 序列兩端會先銜接上一段序列 (adapter) 使其形成環狀 DNA。透過 PacBio 定序系統獨有的循環共識定序模式 (circular consensus sequencing, CCS),待測 DNA 將會被反覆測序。藉由比對反覆測序的序列結果,定序錯誤將得以獲得修正,最終達到 >99.9% (QV30) 的高準確度。 IMAGE © PacBio 2020 [2]. PacBio HiFi 16S 全長定序提供更精確的微菌叢分析結果 在進行微生物體研究時,通常會使用 16S rRNA 或 18S rRNA 基因來鑑定待測樣本中的微菌叢是由哪些成員所組成;其中 16S rRNA 基因用於細菌、古菌、藻類分類鑑定,18S rRNA 基因則用於真菌。 以細菌鑑定為例,目前最為普遍的方式就是使用次世代定序 (next-generation sequencing, NGS) 技術檢測 16S rRNA 基因 9 個可變區 (variable region) 的某個(例如 V4)或某段區域(例如 V1-V3)的 PCR 產物序列,再透過與基因資料庫進行序列比對,最終以序列相似度排列完成分類。此種細菌鑑定方式的解析度可達到「屬 (genus)」的層級。 然而美國傑克森基因體醫學實驗室 (The Jackson Laboratory for Genomic Medicine) Jethro S. Johnson 等人已證實僅使用 16S rRNA 部分可變區序列資訊無法通盤有效地對細菌物種進行分類,會隨著所選用的可變區而有分類效能上的偏向性。唯有使用 16S rRNA 基因全長序列(涵蓋 V1-V9),才能獲得精確無偏差的細菌分類結果(圖 3)[3]。
圖 3﹑唯有使用 16S rRNA 基因全長序列,才能獲得精確無偏差的細菌分類結果。以部分或完整的 16S rRNA 基因序列進行細菌物種分類,紅色代表未能被分類的細菌物種比例,顏色愈深代表該序列的分類效果愈差。從圖中可看出,全長序列分類效果最佳,V4 區域則最差。 IMAGE © Nat Commun. 2019 Nov 6;10(1):5029. Fig. 1c [3]. 使用 PacBio HiFi 定序技術檢測 16S rRNA 基因全長序列,可將細菌物種分類解析度提高至「種 (species)」的層級;並且由於 HiFi 定序數據具有極高的準確度,藉由分析 16S rRNA 基因的單一核苷酸變異 (single-nucleotide polymorphisms, SNPs) 圖譜,還可將分類解析度進一步提升至「株 (strain)」的層級 [3, 4]。目前 HiFi 定序技術已成熟地應用在包含模擬菌群 (mock community) [2-5]、糞便 [3, 4]、湖水 [5]、廢水處理厭氧消化反應器污泥 [6] 等多種類型樣本的 16S rRNA 基因全長定序。
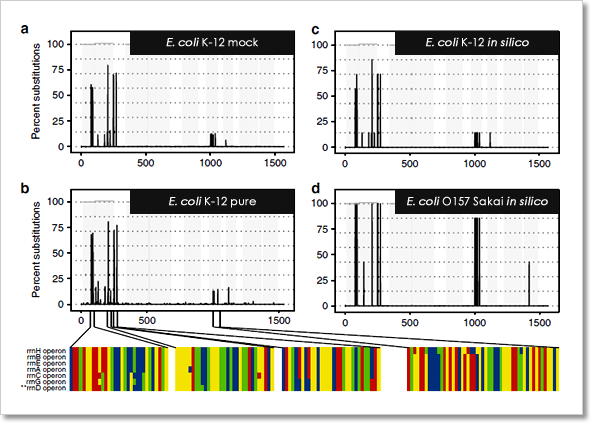
圖 4﹑PacBio HiFi 定序技術的高準確度,讓您可以藉由分析 16S rRNA 基因全長序列的 SNPs 發生位置與機率,將分類解析度提高至「株 (strain)」的層級。圖中所示 16S rRNA 基因 SNPs 分析圖譜分別來自 (a) 模擬菌群中的 E. coli K-12 菌株,採用 PacBio HiFi 定序數據;(b) 實驗室培養分離的 E. coli K-12 菌株,採用 Illumina 定序數據;(c) E. coli K-12 菌株,採用資料庫中的參照基因序列比對而來;(d) E. coli O157 Sakai 菌株,採用資料庫中的參照基因序列比對而來。 IMAGE © Nat Commun. 2019 Nov 6;10(1):5029. Fig. 2 [3].
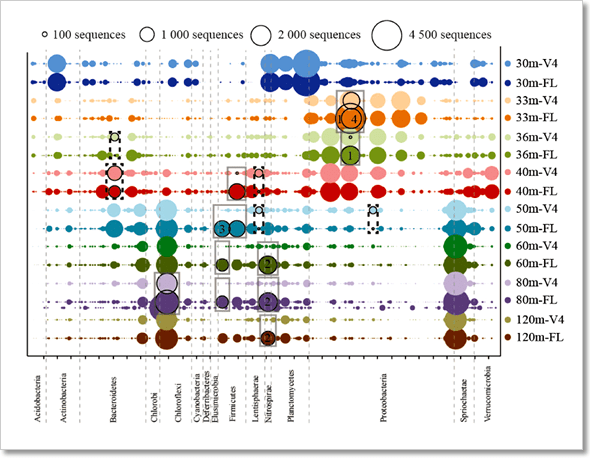
圖 5﹑PacBio HiFi 16S 全長定序讓您發現更加豐富的微生物多樣性。使用 16S rRNA 基因全長 (FL) 序列(PacBio)與 V4 區域(Illumina)定序結果繪製出加拿大薩克內沃湖 (Sakinaw Lake) 不同深度湖水樣本的微生物群落結構示意圖。從圖中可以觀察到,許多優勢物種(灰色方框處)唯有透過 16S rRNA 基因全長定序方式才能被解析出來。 IMAGE © ISME J. 2016 Aug;10(8):2020-32. Fig. 4b [5]. PacBio HiFi 總體基因體定序 = 發現更多基因 + 組裝更容易 總體基因體 (metagenome) 是微菌叢所有組成成員的基因體合集 [1],透過 PacBio HiFi 定序技術進行總體基因體定序 (shotgun metagenomic sequencing),可以幫助科研人員取得更精準的 16S 菌群分類結果(因為總體基因體定序不經過 PCR 過程,可以避免掉 PCR 反應可能帶來的偏差效應)[5, 7];同時由於 HiFi 定序數據兼具有長讀取與高準確度的特性,相較於短讀取定序技術與容易產生錯誤的其他長讀取定序技術,HiFi 定序數據能夠讓科研人員更容易地從 Contig 中發掘出基因與操縱子 (operon),也讓代謝路徑探索與序列組裝工作變得更有效率 [2, 8-10]。
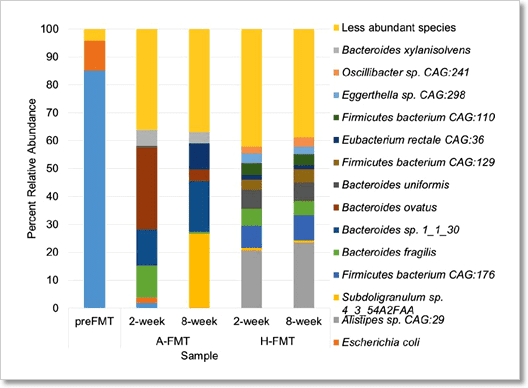
圖 6﹑PacBio HiFi 定序技術能夠精確地鑑別出微菌叢植入 (fecal microbiota transplantation, FMT) 病患腸道微生物群落結構變化,解析度可達「種 (species)」的層級。使用 PacBio HiFi 定序技術檢測糞便檢體的總體基因體,並依據序列分析結果繪製出腸道微生物群落結構示意圖。糞便檢體分別來自接受 FMT 治療前的病患(preFMT)、接受自體糞便 (autologous FMT) 治療後的病患(A-FMT)以及接受捐贈者糞便 (heterologous FMT) 治療後的病患(H-FMT)。從圖表中可看出,H-FMT 治療方式能夠提供更長期穩定的菌群改善效果。 IMAGE © Gut Microbes. 2017 May 4;8(3):268-275. Fig. 4 [7].
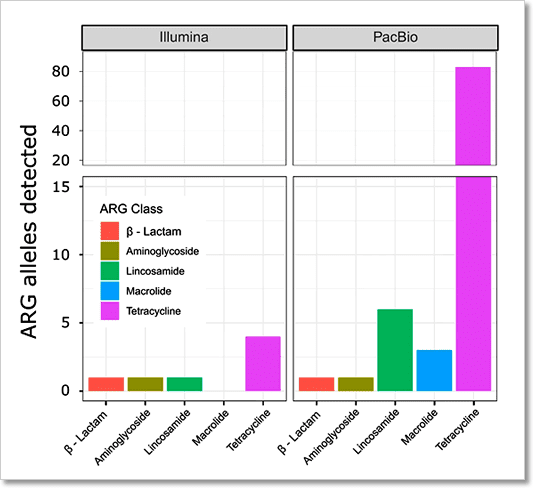
圖 7﹑使用 PacBio HiFi 技術進行總體基因體定序,讓您發現更多基因。分別使用 PacBio HiFi 與 Illumina 定序技術進行乳牛瘤胃 (rumen) 樣本的總體基因體定序。相較於 Illumina,PacBio HiFi 定序可以鑑定找尋出更多的抗藥性基因 (antimicrobial resistance gene),其數量為 Illumina 的十倍以上。 IMAGE © Genome Biol. 2019 Aug 2;20(1):153. Fig. 5b [8].
圖 8﹑使用 PacBio HiFi 技術進行總體基因體定序,讓序列組裝更容易。加州大學 Mikhail Kolmogorov 等人利用專為總體基因體序列組裝所開發的 metaFlye 軟體,成功地自羊腸道微菌叢的 PacBio HiFi 總體基因體定序結果中組裝出 63 個完整或近乎完整的細菌基因體。本圖為梭菌綱 (Clostridia class) 的組裝示意圖。 IMAGE © Nat Methods. 2020 Nov;17(11):1103-1110. Fig. 4a [9]. 由 PacBio 微生物部門總監 Meredith Ashby 所主講的這個短片,將能夠帶領您快速了解 HiFi 定序技術在總體基因體學 (metagenomics) 研究的卓越效能與潛力。完整產品資訊與最新活動訊息,歡迎洽詢 PacBio 台灣代理 — 伯森生技。 References
|
|

伯森生物科技(股)公司 Blossom Biotechnologies, Inc.
網址 www.blossombio.com 客服 0800-059668
[ 📝 線上留言諮詢 ] [ ☎ 伯森業務專員聯絡資訊 ]
![]()
![]()